Федеральное казенное учреждение. государственное (муниципальное) казенное учреждение, его деятельность и права
Содержание:
- Психологические тенденции
- Фенилкетонурия и материнство
- Симптомы заболевания
- Прогноз и профилактика
- Диагностика фенилкетонурии
- Генотерапия
- Примеры из жизни
- Клиническая картина
- Создание казенного учреждения
- К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
- Лечение фенилкетонурии
- Симптомы фенилкетонурии
Психологические тенденции
Интеллектуальное развитие было измерено при помощи шкалы Wechsler Adult Intelligence Scale-Revised (WAIS-R). На графике 1 видны баллы в зависимости от возраста, в котором была прекращена диета: первая группа слева направо – диета прекращена до достижения 6.5 лет, вторая – диета прекращена в возрасте 6.5-12.5 лет, третья – диета прекращена в возрасте 12.5-20 лет, четвертая – диета никогда не прекращалась. Из этого графика ясно видно, что пациенты, остававшиеся всю жизнь на диете, имели значительно более высокое интеллектуальное развитие, в том числе и сравнивая их показатели с ожидаемым уровнем, основанным на IQ баллах их родителей.
Примите во внимание, что при анализе этих баллов группа пациентов, которая постоянно находилась на диете и имела в среднем уровень фенилаланина в крови выше оптимального (15 mg/dl или 926 .mmol/L, и у нескольких пациентов – значительно выше), и средний уровень фенилаланина к крови в детстве около 10 mg/dl. Тем не менее IQ баллы группы, которая находилась на диете, были в пределах 3 баллов от показателей их родителей, в то время как в группе пациентов сошедших с диеты IQ баллы были гораздо ниже чем у их родителей
Когда баллы в зрелом возрасте были противопоставлены баллам в возрасте 12 лет, было замечено значительное повышение в IQ для группы никогда не прекращавшей диету. Статистический анализ показал сильную зависимость между IQ баллами во взрослых пациентах и IQ родителей, уровнем их образования, возраста, в котором была начата диета, возраста в котором диета прекратилась, а так же уровень фенилаланина в крови в разном возрасте.
Иными словами – чем выше IQ родителей, их образование, чем раньше ребенок начал диету, чем ниже были уровни фенилаланина в крови на протяжении соблюдения диеты и продолжение деты в течение всего срока – все эти факторы имели благотворное влияние на IQ взрослых пациентов. Из 16 взрослых пациентов с классической формой фенилкетонурии, которые вернулись на диету, 9 продолжали пить гидролизат и имели более высокие IQ баллы чем в детстве. В то же время, 7 пациентов, которые прекратили диету продемонстрировали тенденцию снижения IQ баллов во взрослом возрасте, по сравнению с детским возрастом. Что интересно – пациенты с классической формой фенилкетонурии которые на данный момент исследования пили гидролизат имели значительно более высокие показатели IQ чем пациенты на обычной диете, даже если их уровень фенилаланина в крови был выше нормы.
Фенилкетонурия и материнство
Для беременных женщин, больных ФКУ, очень важно удерживать низкий уровень фенилаланина до и во время всей беременности, для того, чтобы ребенок был здоровым. И хотя, развивающийся плод может быть только носителем гена ФКУ, однако внутриутробная среда может иметь очень высокий уровень фенилаланина, который обладает способностью проникать через плаценту
Как следствие, у ребенка может развиваться врожденный порок сердца, возможна задержка развития, микроцефалия и умственная отсталость. Как правило, у больных фенилкетонурией женщин никаких осложнений во время беременности не возникает.
Именно поэтому, ежедневное количество потребленного матерью фенилаланина может удвоиться или даже утроиться к концу беременности. Если же уровень фенилаланина в крови матери ниже 2 мкмоль / л, то иногда, у женщин могут возникать различные осложнения, связанные с дефицитом этой аминокислоты, такие как головная боль, тошнота, выпадение волос и общее недомогание. Если низкий уровень фенилаланина у больных ФКУ поддерживается в течение всей беременности, то риск родить больного ребенка не выше, чем у тех женщин, которые не больны ФКУ.
Симптомы заболевания
При своевременном обнаружении болезнь Феллинга поддается успешному лечению путем корректировки питания, и развитие ребенка происходит в соответствии его возрастной группе. Трудность выявления генной мутации заключается в том, что ранние признаки тяжело обнаружить даже опытному педиатру. Выраженность симптоматики врожденного заболевания усиливается по мере взросления ребенка, потому что употребление белковой пищи способствует развитию нарушений ЦНС.
Признаки у новорожденных
На протяжении первых дней жизни ребенка признаки патологических отклонений обнаружить трудно – малыш ведет себя естественно, задержки в развитии не наблюдается. Симптомы заболевания впервые начинают проявляться через 2-6 месяцев после рождения. Родителей должно насторожить поведение малыша, которое характеризуется низкой активностью, вялостью, или, наоборот, беспокойством, гипервозбудимостью.
С началом грудного вскармливания в организм новорожденного с молоком начинают поступать белки, что служит катализатором появления первых признаков, однозначно свидетельствующих о том, что заболевание начало прогрессировать. К специфическим клиническим проявлениям болезни относятся:
- постоянная рвота (зачастую принимаемая за врожденное сужение привратника);
- частое срыгивание;
- отсутствие реакции на внешние раздражители;
- мышечная дистония (сниженное напряжение мышц);
- судорожный синдром (судороги эпилептического или неэпилептического характера).
Симптомы у детей после 6 месяцев
Если манифестация генетической болезни не произошла (или не была замечена) в течение первых 6 месяцев с момента рождения ребенка, то после этого периода уже можно точно определить отставание в психомоторном развитии. Симптомами генетических нарушений, вызванных ферментным дефицитом, у детей, старше полугода, являются:
- снижение активности (вплоть до полной безучастности);
- отсутствие попыток к самостоятельному вставанию, сидению;
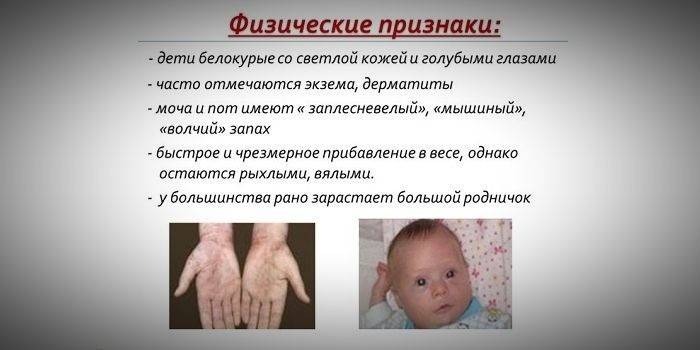
- особенный «мышиный» запах кожи (запах плесени возникает вследствие выведения токсических производных фенилаланина через потовые железы и мочу);
- потеря способности к визуальному распознаванию лиц родителей;
- шелушение кожи;
- появление дерматитов, экзем, склеродермии.
Прогрессирование заболевания при отсутствии лечения в детском возрасте
Если отклонения в развитии не были выявлены в младенческом возрасте, и соответствующее лечение не проводилось, то заболевание начинает активно прогрессировать и нередко приводит к инвалидности. Отсутствие терапии на раннем этапе болезни вызывает появление следующих симптомов болезни уже в возрасте 1,5 лет:
- микроцефалия (уменьшенные размеры головного мозга);
- прогнатия (смещение верхнего зубного ряда вперед);
- позднее прорезывание зубов;
- гипоплазия эмали (истончение или полное отсутствие зубной эмали);
- задержка речевого развития вплоть до полного отсутствия речи;
- 3, 4 степень олигофрении (задержка психического развития, умственная отсталость);
- врожденные пороки сердца (дефекты в структуре сердечной мышцы, отделах сердца, крупных сосудах);
- расстройства вегетативной системы (акроцианоз, повышенная потливость, артериальная гипотония);
- запоры.
Прогноз и профилактика
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.
https://youtube.com/watch?v=pww9ml5pywE
Диагностика фенилкетонурии
 Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия — самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия — самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Скрининг-тест производится не раньше чем на 3 сутки (72 часа) после рождения ребенка. При обнаружении у ребёнка повышенного уровня фенилаланина (гиперфенилаланемии) скриниговая лаборатория согласовывает тактику в отношении каждого пациента с ведущим врачом-генетиком.
При уровне ФА в скриниге:
- 3-8мг% — родителей или опекунов информируют о необходимости выполнения контрольного исследования (колориметрическим методом). Если в котрольном исследовании уровень ФА выше 2мг%, то необходимо оповестить родителей/опекунов и пригласить в письменной форме в специализированный диагностический лечебный центр;
- выше 8мг% — необходимо оповестить родителей/опекунов о результате скринигового теста и пригласить в письменной форме в специализированный диагностический лечебный центр.
Во время первого визита врач-специалист (чаще всего педиатр или генетик) обязан предоставить родителям/опекунам исчерпывающую информацию относительно предполагаемого заболевания, его причин и возможностей лечения.
В связи с существованием ряда причин появления гиперфенилаланемии (фенилкетонурия, нетипичные формы фенилкетонурии и тирозенемия), необходимо провести дифференциальную диагностику. Если подтвердится дефицит гидроксилазы фенилаланина (фенилкетонурия), применяется диетотерапия. В случае, если причиной окажется нарушение обмена веществ иного рода, будут избраны иные методы лечения.
Типы фенилкетонурии
Классическая фенилкетонурия –phe > 1200 μмол/л (20 мг%) Умеренная фенилкетонурия – phe – 900 -1200 μмол/л (15 — 20 мг%) Мягкая фенилкетонурия – phe –600–900 μмол/л (10-15мг%) Мягкая гиперфенилаланинемия (gray zone) — phe – 360 – 600 μмол/л (6 — 10мг%) Мягкая гиперфенилаланинемия- не нуждающаяся в лечении – phe – 120 – 360 μмол/л (2 — 6мг%) (NIH PKU Conference report: State of the science and future research needs. Feb.22-23.2012) Злокачественная гиперфенилаланинемия – недостаточность тетpагидpобиоптеpина (BH4)
Дефицит гидроксилазы фенилаланина (PAH)
— фенилаланин ≤ 7 мг% (6 мг%) не нуждается в лечении – наблюдение!- фенилаланин > 7 мг% (6 мг%) – низкофенилаланиновая диета
Дефицит BH4 (злокачесивенная ФКУ) — фармакологическое лечение.
Клинические появления злокачественной фенилкетонурии:
— тяжелые и быстро прогрессирующие неврологические нарушения (несмотрия на нормальный уровень ФА в крови вследстве диетотерапии)- гипотония- спастический синдром- атаксия- нарушение глотания- беспокойство- прогрессирующая умственная отсталость- тяжело купирующиеся приступы судорог – наиболее характерные (до 3 мес. жизни признаки заболевания могут отсутствовать)- уровни фенилаланина нехарактерные ( высокие – как в классической ФКУ или низкие — как в мягкой гиперфенилаланинемии).
Прогрессирующее повреждение нервной системы при злокачественной фенилкетонурии может вести к смерти ребенка, если заболевание не будет обнаружено и не будет применено соответствующее лечение.
Также о дифференцированной диагностике читайте в статье Рекомендации по лечению фенилкетонурии в Польше
Генотерапия
Кроме диетотерапии, возможен альтернативный путь лечения фенилкетонурии путем генотерапии.
Из 3-х основных шагов, требуемых для генотерапии при фенилкетонурии, два выполнены: получена кДНК, обеспечивающая экспрессию гидроксилазы фенилаланина человека, разработана гидроксилаза-дефицитная животная модель.
Векторы для эффективной передачи гена in vivo требуют дальнейшей разработки.
Ретровирусные векторы, несмотря на то, что достаточно эффективны in vitro, имеют низкую эффективность передачи in vivo.
Рекомбинантные аденовирусные векторы, хотя и полностью успешны на короткое время, не сохраняются в организме больше нескольких недель из-за иммунного ответа.
Примеры из жизни
Под наблюдением Кабинета Фенилкетонурии в детской больнице Кракова в 2009 году находилось 430 пациентов с гиперфенилаланинемией в возрасте от 0 до 56 лет, в том числе 81 женщина старше 16 лет.
Ретроспективно проанализированы все беременности с 1985 по 2009 год
-
- классической фенилкетонурией (ФА без диетического контроля >20mg/dl)
- умеренной фенилкетонурией (ФА без диетического контроля 10-20mg/dl) и
- Умеренной гиперфенилаланинемией (ФА без диетического контроля 6-10mg/dl) старше 16 лет
-
Оценке подвергался диетический контроль у женщин с PKU/HPA:
- в пре- и
- постконцепционном периодах
-
Хорошим признавался контроль, если удавалось достичь:
- стабилизации концентраций ФА в сыворотке крови в границах 2-6mg/dl в преконцепционном периоде и
- сохранить такой уровень контроля в течение всей беременности, a особенно в I и II триместрах
-
10/50 беременностей наших пациенток были планированы
-
диета с низким содержанием фенилаланина внедрена 3 месяца перед и ведена на протяжении всей беременности с целью сохранения уровня контроля в границах от 2,0 до 6,0mg/dl
-
1/10 беременностей закончились самопроизвольным выкидышем
-
9/10 беременностей закончились рождением здорового ребенка
-
К сожалению 40/50 были беременностями непланированными: диета не соблюдалась в периоде пре- и посткоцепционном или была введена поздно
-
8/40 беременностей закончились самопроизвольным выкидышем
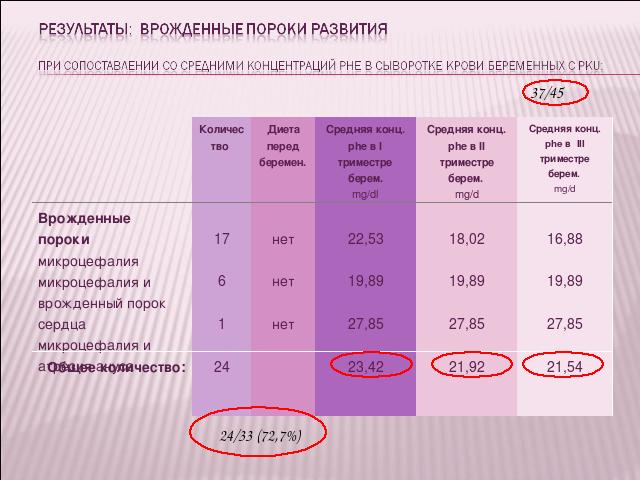
- а остальные 32/40 беременностей закончились рождением 24/33 (72,7%) с синдромом материнской ФКУ.
Результаты
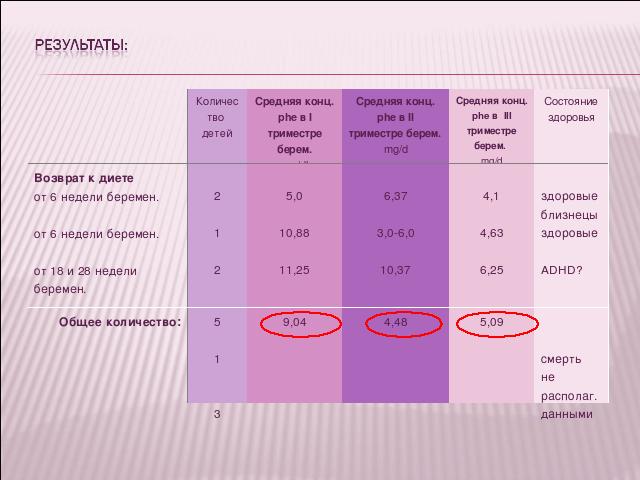
Восемь беременных возобновили диету поздно (6-28 нелдели), уровни ФА заранее не контролировались, а при поступлении на учет значительно превышали нормальные. Введденная даже в этом периоде терапия у четырех из десяти пациенток с ФКУ способствовала рождению 5 (15, 79%) клинически здоровых детей.
Пациентка К.К., 17 лет
— диагностировано ФКУ в скрининге новорожденнных
— под опекой Метаболического Центра от момента установления диагноза
— диетическое лечение внедрено в соответствующем периоде, но …
— в подростковом возрасте становилось все более либеральным, контрольные концентрации ФА были очень высокими
— оценка интеллектуального развития на нижней границы возрастой нормы
— недостаточно добросовестное сотрудничество со стороны родителей, отсутствие акцептации болезни окружающими.
Появилась в Клинике на 12 неделе беременности, концетрация ФА, определенная в этом периоде — 20,5мг%. Немедленно при содействии с мужем пациетки внедрено ригористическую диету, что позволило получить метаболический эффект.
K.K. родила дочь без признаков синдрома материнской PKU, но остается под постоянным наблюдением по поводу синдрома ADHD.
Пациентка Б.Г., 30 лет
Сдиагнозирована в периоде новорожденности при помощи пилотажного скрининга, проводимого Институтом Матери и Ребенка в Варшаве до 1985 года. Лечение внедрено от момента установления диагноза – перед окончанием 3 месяца жизни
В нашем наблюдении от 03.08.1993 года
Из анамнеза:
берем. I. самопроизвольный аборт
берем. II, ребенок с синдромом материнской фенилкетонурии — микроцефалией
берем. III, ребенок с синдромом материнской фенилкетонурии — микроцефалией
берем. IV, самопроизвольный аборт
От 1999 года у пациентки наблюдаются невро-вегетативные нарушения, депрессия, в связи с чем остаётся в постоянном неврологическом и психиатрическом лечении
берем. V, явилась на 6 неделе.
Клиническая картина
Для больных фенилкетонурией характерен светлый цвет волос и голубая радужная оболочка глаз (дети всегда светлее своих родителей и здоровых братьев и сестер). В первые 2_3 месяца жизни большинство детей выглядит совершенно здоровыми. Лишь у некоторых из них отмечается вялость, сонливость, реже беспокойство. Ранним симптомом является рвота. Отмечается своеобразный запах мочи ребенка (затхлый, мышиный). К концу первого полугодия жизни у 20—50% больных детей появляются экзематозные изменения кожи, иногда значительные, у некоторых развивается склередема (см. Склередема новорожденных). В этом же возрасте начинает выявляться задержка психического развития, к-рая, нарастая, в дальнейшем приводит к тяжелому слабоумию (см.) — имбецильности или идиотии. Лишь у незначительного числа больных поражение интеллектуальной сферы выражено нерезко. Отмечается отставание в физическом развитии. Задерживается формирование двигательных навыков. У многих больных появляются судороги, которые могут постепенно исчезать даже без лечения, обнаруживается гилеррефлексия, непроизвольные движения. У большинства больных на ЭЭГ регистрируется пароксизмальная активность. У некоторых детей отмечается спазм мышц конечностей (преимущественно ног). Все отмеченные изменения характерны для больных, не получающих с рождения патогенетического лечения.
Создание казенного учреждения
Такой тип государственных учреждений создается по распоряжению федерального или регионального органа исполнительной власти, муниципалитета (ч. 2 ст. 13 закона № 7-ФЗ).
Обратите внимание! На федеральном уровне этот процесс регламентирован Порядком создания, реорганизации, изменения типа, ликвидации…, утв. постановлением Правительства РФ от 26.07.2010 № 539
На уровне субъекта РФ или муниципалитета действуют уже свои акты.
Пример — Порядок, утв. постановлением правительства Тюменской области от 28.12.2010 № 393-П, и Порядок, утв. постановлением администрации Заводоуковского городского округа от 30.11.2010 № 1887.
Обычно процесс создания казенного учреждения включает следующие этапы:
- Принятие учредителем решения о создании юрлица.
- Оформление проекта распоряжения/постановления о создании учреждения. Он, как правило, содержит:
- название учреждения, его тип;
- основные виды и цели деятельности, функции казенного учреждения, разрешенные для данного казенного учреждения платные услуги и т. д.;
- наименование учредителя;
- перечень мероприятий, направленных на создание учреждения, с обозначением их сроков и ответственных за их реализацию структур.
- Составление пояснительной записки к проекту с обоснованием целесообразности создания учреждения.
- Согласование проекта с ответственными подразделениями учредителя (например, департаментом имущественных отношений и правовым департаментом).
- Издание постановления о создании учреждения.
- Утверждение устава учреждения путем издания распоряжения/ постановления.
- Направление документов в ФНС для государственной регистрации юрлица.
Рекомендуем! Как направить документы для госрегистрации юрлица через портал «Госуслуги», читайте в нашей статье «Регистрация юридического лица на портале госуслуг».
Устав
Учредительный документ казенного учреждения — устав — содержит сведения:
- о названии юрлица, его типе (в названии учреждения обязательно должно быть указано, что оно казенное);
- местонахождении юрлица;
- учредителе и собственнике имущества;
- видах деятельности, организации, управлении юрлицом, его структуре и т. п.;
- имуществе и финансовом состоянии, в т. ч. порядке распоряжения имуществом, его передаче другим учреждениям, совершении крупных сделок и сделок с заинтересованностью и т. д.;
- обособленных подразделениях юрлица и т. д.
Обратите внимание! Устав утверждает учредитель юрлица посредством принятия правового акта. Порядок согласования, утверждения и внесения изменений в устав обычно регламентирован
Например, для устава федерального государственного казенного учреждения порядок утвержден постановлением Правительства РФ от 26.07.2010 № 539.
Руководитель
Единоличный исполнительный орган управления казенного учреждения — директор/руководитель. Его назначает собственник имущества учреждения, издав специальное распоряжение. Директора выбирают посредством проведения конкурса. Он подотчетен собственнику имущества.
Директор выполняет функции по организации деятельности учреждения в соответствии с целями последнего, совершает сделки от его имени, решает кадровые, административно-хозяйственные и прочие вопросы, возникающие в процессе работы юрлица.
Имущество казенного учреждения
Рекомендуем! О том, как зарегистрировать право оперативного управления на имущество, читайте в нашей статье «Государственная регистрация права оперативного управления на объекты недвижимости».
К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
Педиатр
Психоневролог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Фенилкетонурии (ФКУ) у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог. Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен. В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги. После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится. Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.